

Wirus PCV2 pojawił się jako czynnik wywołujący jedną z najgroźniejszych chorób wirusowych trzody chlewnej będącą przyczyną dużych strat ekonomicznych związanych z chorobą i kosztami jej zwalczania, kiedyś nazwanej zespołem wyniszczenia poodsadzeniowego świń (PMWS), a obecnie określanej mianem uogólnionej (systemowej) choroby związanej z PCV2 (PCV2-SD, ryc. 1). Badania epidemiologiczne i doświadczalne wykazały, że zmienność genetyczna może potencjalnie wpływać na zjadliwość PCV2, co może uwidaczniać się w postaci pojawiania się i globalnego rozprzestrzenienia nowych, np. rekombinowanych szczepów wirusa. Sprawia to również trudności w klasyfikacji wariantów PCV2. W 2008 roku, konsorcjum projektu poświęconego badaniom chorób związanych z PCV2 zaproponowało nomenklaturę genotypów PCV2 opartą na porównaniu sekwencji nukleotydowych (www.pcvd.eu). Analizy kompletnego genomu i genu kodującego białko kapsydu (ORF2) pozwoliła na określenie dwóch wartości granicznych dystansu genetycznego, odpowiednio 0,020 i 0,035. Jeśli dystans między dwoma różnymi sekwencjami (całego genomu lub ORF2) jest większy od tej wartości to należy to interpretować to w ten sposób, że mamy do czynienia z dwoma różnymi genotypami. Wynikiem badań było wyróżnienie czterech genotypów PCV2: PCV2a, PCV2b, PCV2c i PCV2d (znanego również jako mutant PCV2b).


Ryc. 1. Trzymiesięczna świnia z objawami PCV2-SD. Widoczne wychudzenie, zahamowanie wzrostu i bladość powłok skórnych (anemia).
Od 2008 roku w GenBank (www.ncbi.nlm.nih.gov) zdeponowano olbrzymią liczbę sekwencji PCV2, oraz zaproponowano istnienie kilku nowych genotypów, co jednak nie zawsze były odpowiednio uzasadnione. Duża ilość tych sekwencji pochodzi od zrekombinowanych wirusów a w niektórych przypadkach zauważono, że występowanie takich szczepów rosło w krajach azjatyckich i USA. W publikacji Franzo i wsp. z 2015 roku opisano wyniki badań międzynarodowego zespołu, który przeprowadził powtórną analizę znanych sekwencji PCV2 w celu unifikacji nomenklatury szczepów i uniknięcia przyszłych błędów w genotypowaniu.
Uzyskane wyniki wykazały, że analiza dostępnych obecnie sekwencji kapsydu i kompletnych genomów PCV2 przy użyciu metodologii sprzed około dekady prowadziła do uzyskania nieprawidłowych danych. Błędne było założenie równego współczynnika podstawień nukleotydowych u różnych genotypów. Okazało się również, że zastosowanie jednej wartości odcięcia dystansów genetycznych jest trudne. Wcześniej ustalone wartości okazały się obecnie zupełnie niewłaściwe (ryc. 2). Dowody te wraz z dostępnością olbrzymiej liczby nowych sekwencji mają istotne znaczenie dla klasyfikacji PCV2, i wymagają powtórnego jej opracowania.
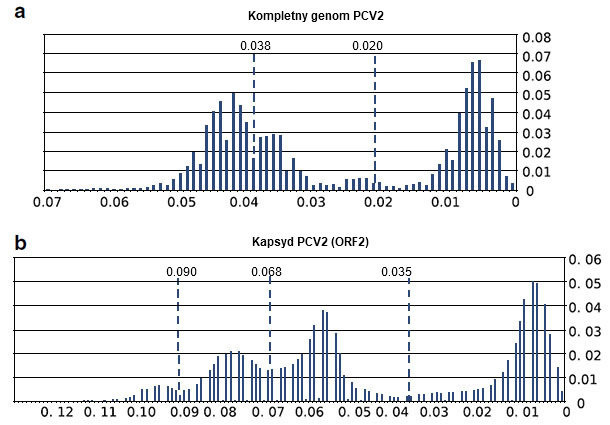
Ryc. 2. Analiza zmienności kompletnych genomów PCV2 (a) i ORF2 (b). Pokazano proporcje dystansów między parami sekwencji zawartych w interwałach dystansów co 0,01. Pokazano również proponowane wartości graniczne dla kompletnych genomów (0,020), ORF2 (0,035) oraz wartości po powtórnych analizach (0,038, 0, 068 i 0,090).
Zgodnie z tym zaproponowano nową, alternatywną, w większym stopniu jednoznaczną metodę genotypowania PCV2. Metoda ta bierze pod uwagę obecność rekombinantów należących do kilku genotypów. Biorąc pod uwagę wysoką częstość występowania rekombinacji do genotypowania szczepów wybrano metodę oparta na analizie ORF2. Najpierw wybrano szereg sekwencji, których klasyfikacja była jednoznaczna. Pozwoliło to na zdefiniowanie czterech genotypów na podstawie analizy filogenetycznej jak i markerów genetycznego na 47 pozycjach nukleotydowych. Te pozycje markerowe pozwalają w >95% na prawidłową klasyfikację szczepów w genotypy. Zaproponowana metoda klasyfikacji jest szybka i dokładna.
Klasyfikacja szczepów PCV2 stale sprawia trudności ze względu na rekombinację do jakiej dochodzi między wirusami należącymi do jednego jak i różnych genotypów. Ponadto cały czas istnieje prawdopodobieństwo pojawienia się nowych genotypów PCV2 i nowe narzędzia genotypowania mogą okazać się niezbędne.
Klasyfikacja genotypów PCV2 jest ważna ze względów praktycznych. Jak na razie wszystkie dostępne szczepionki w Europie i Ameryce Północnej są opracowane w oparciu o genotyp PCV2a, podczas gdy najczęściej spotykane w terenie szczepy należą do PCV2b i PCV2d. Mimo, że poziom odporności krzyżowej między tymi trzema genotypami jest wysoki, byłoby interesującym zbadać czy skuteczność szczepionki jest jednakowa w odniesieniu do wszystkich genotypów. Ponadto kilka badań filogenetycznych wykazało obecność zmienionych aminokwasów u szczepów pochodzących ze szczepionych ferm. Spekuluje się na temat możliwości pojawiania się szczepów-mutantów unikających odporności poszczepiennej. Z powyższych względów genotypowanie PCV2 może być bardzo ważne w ocenie skuteczności szczepionek.

